More cell and gene therapy products are being developed and entering clinical trials each year. The U.S. Food and Drug Administration plays a key role in overseeing drug development, including providing guidance and receiving investigational new drug applications or requests to start a new clinical trial submitted by drug developers.Â
In 2021, there are more than 1,000 cell and gene therapy clinical trials, including more than a dozen in hemophilia. FDA announced it expects more than 200 new requests to start clinical trials based on gene and cell technologies each year. By 2025, FDA expects 10 to 20 new CGT treatments will be approved annually.Â
Gene therapy treatments for hemophilia have shown potential to eliminate the need for prophylactic factor infusions and injections. Unlike gene therapy, newer technologies such as genetically modified factor-producing cells do not involve changes in patient genetic material and may allow better control of dosing as well as redosing. But many challenges and uncertainties face researchers and drug developers. To help set best practices across the industry, FDA guidance frameworks play an important role in safe and efficient development of CGT products.Â
What are FDA Guidance Frameworks?
To help the companies developing new drugs, FDA publishes recommendations called guidance documents. Unlike the laws passed by Congress or formal regulations, guidance  documents contain FDA expectations and current scientific thinking not required by law. If we think of regulations as what the law requires drug developers to do, we can think of guidance documents as a playbook of how to do it in most, but not all, cases.
documents contain FDA expectations and current scientific thinking not required by law. If we think of regulations as what the law requires drug developers to do, we can think of guidance documents as a playbook of how to do it in most, but not all, cases.
Guidance frameworks are groups of related guidance documents, such as those relating to CGT. New guidance creation is an important part of FDA’s ongoing mission to expedite innovations that make medical products more effective, safer and more affordable. Guidance documents are written and published according to a process called Good Guidance Practice, which describes how FDA staff will bring together expert and public feedback in guidance recommendations.
Growing Expectations for CGT in Hemophilia
FDA’s first draft guidance document dedicated to early development of CGT clinical trials – Guidance for Industry: Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products – was released as a draft in 2013 and finalized in 2015. It discussed many recommendations for manufacturing, testing and patient safety and follow-up.
In 2016, Congress passed the 21st Century Cures Act or Cures Act, which includes provisions designed to expedite and streamline development of innovative new medicines. This law builds on FDA’s existing responsibilities and established regenerative medicine therapies, including gene therapy, cell therapy, products made from tissues and combinations of these products. FDA published guidance in 2017 and updated in 2020 to help industry apply these new regulations that together are referred to as the FDA regenerative medicine framework. Both human gene editing and transfer, as well as genetically modified cells that lead to a sustained factor production in hemophilia, are considered to be regenerative medicine therapies by FDA.
Guidance for developing CGT products specifically for hemophilia was published by FDA in 2018. Then in 2020, it was updated when FDA launched its expanded framework for cell and gene therapies. This framework contained several new guidance documents, including hemophilia CGT. This brought the total number of guidance documents in the framework to 27. In 2021, we can see how the evolution of hemophilia gene and cell therapy has been affected by the development of this framework.Â
GUIDANCE, REGULATIONS AND LAWS THAT MAKE UP THE U.S. FDA FRAMEWORK FOR DEVELOPING NEW CELL AND GENE THERAPY (CGT) PRODUCTS FOR HEMOPHILIA
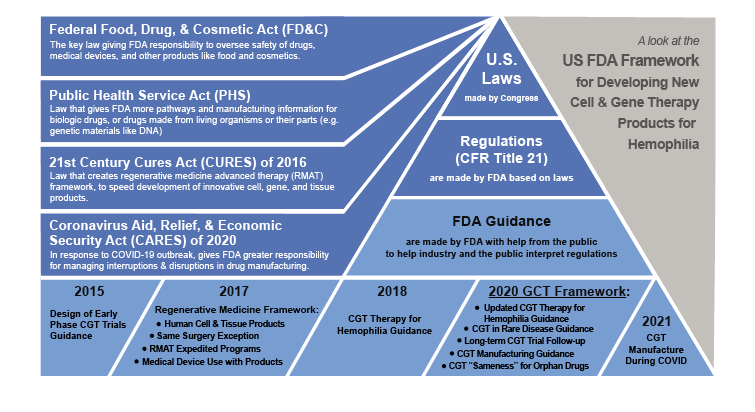
Early 2020: FDA Launches Cell and Gene Therapy Guidance Framework
On Jan. 28, 2020, FDA launched its landmark guidance framework for CGT products. Updates included improvements to the guidance for hemophilia and rare disorders first drafted in 2018, including Guidance for Industry: Human Gene Therapy for Hemophilia and Guidance for Industry: Human Gene Therapy for Rare Diseases. FDA also confirmed draft  recommendations, Guidance for Industry: Long Term Follow- Up After Administration of Human Gene Therapy Products, to follow patients for safety for five to 15 years after participating in a CGT clinical trial. Â
recommendations, Guidance for Industry: Long Term Follow- Up After Administration of Human Gene Therapy Products, to follow patients for safety for five to 15 years after participating in a CGT clinical trial. Â
The framework also updated guidance for best practices for manufacturers to make and test new CGT products, Guidance for Industry: Chemistry, Manufacturing, and Control Information for Human Gene Therapy, Investigational New Drug Applications. Finally, a new draft guidance was published to help clarify that products treating rare disease must be unique (i.e., not “sameness” such as minor variations of the same cell or genetic material) to apply for orphan drug status, which gives certain incentives like tax credits, FDA fee waivers and rights to market a new drug exclusively (or without competitors selling the same drug) for seven years after approval, Guidance for Industry: Interpreting Sameness of Gene Therapy Products Under the Orphan Drug Regulations. This guidance framework clarified many of the issues facing developers of cell and gene treatments for hemophilia and other rare bleeding disorders.
The New Landscape for Hemophilia CGT
In 2021, more than a dozen gene transfer, gene editing and genetically modified cell therapy trials for hemophilia are ongoing, with AAV gene therapies the furthest along though lentivirus gene therapy and non-viral genetically modified cell therapies clinical trials. The outbreak of the respiratory disease COVID-19 caused by the novel coronavirus SARS-CoV-2, and passage of the Coronavirus Aid, Relief, & Economic Security Act of 2020 by Congress has shifted the focus of FDA. In January 2021, FDA added recommendations for CGT manufacturers to its guidance framework, Guidance for Industry: Manufacturing Considerations for Licensed and Investigational Cellular and Gene Therapy Products During COVID-19 Public Health Emergency.
CGT for Hemophilia in 2021 and Beyond
Despite many challenges for ongoing clinical trials in 2020, CGT treatments for hemophilia are much closer to reaching patients. Many clinical trials have benefited from wide adoption  of digital health and telemedicine in 2020, and from the additional standardization of CGT manufacturing practices under the updated FDA guidance framework.
of digital health and telemedicine in 2020, and from the additional standardization of CGT manufacturing practices under the updated FDA guidance framework.
The CGT community has taken important early steps on the path to harmonization or development of international recommendations applicable across the world. However, several hemophilia gene therapy trials have also experienced setbacks related to making or distributing CGT products during the COVID-19 outbreak and delays to clinical trials while new scientific and safety information is being investigated and shared with FDA. Such communication between product developers and regulatory agencies, such as FDA, is a very important part of developing safe and effective treatments. The COVID-19 outbreak could also increase the time it takes for these treatments to become widely available. While current guidance framework has helped streamline development of CGT, we expect that FDA will continue to work closely with researchers, product developers and patient groups to further evolve the CGT guidance framework to help ensure that the next generation of hemophilia treatments are developed rapidly, efficiently and safely.Â


