Cada año se desarrollan y entran en ensayos clínicos más productos de terapia celular y génica. La Administración de Alimentos y Medicamentos de EE. UU. desempeña un papel clave en la supervisión del desarrollo de medicamentos, lo que incluye brindar orientación y recibir solicitudes de nuevos medicamentos en investigación o solicitudes para iniciar un nuevo ensayo clínico presentadas por los desarrolladores de medicamentos.A
En 2021, hay más de 1000 ensayos clínicos de terapia celular y génica, incluidos más de una docena en hemofilia. La FDA anunció que espera recibir más de 200 nuevas solicitudes cada año para iniciar ensayos clínicos basados en tecnologías genéticas y celulares. Para 2025, la FDA espera que se aprueben anualmente entre 10 y 20 nuevos tratamientos CGT.A
Los tratamientos de terapia génica para la hemofilia han demostrado tener potencial para eliminar la necesidad de infusiones e inyecciones de factor profiláctico. A diferencia de la terapia génica, las tecnologías más nuevas, como las células productoras de factores modificadas genéticamente, no implican cambios en el material genético del paciente y pueden permitir un mejor control de la dosificación y de la redosificación. Pero los investigadores y los desarrolladores de fármacos enfrentan muchos desafíos e incertidumbres. Para ayudar a establecer las mejores prácticas en toda la industria, los marcos de orientación de la FDA desempeñan un papel importante en el desarrollo seguro y eficiente de productos CGT.A
¿Qué son los marcos de orientación de la FDA?
Para ayudar a las empresas a desarrollar nuevos medicamentos, la FDA publica recomendaciones llamadas documentos de orientación. A diferencia de las leyes aprobadas por el Congreso o los reglamentos formales, la orientación  Los documentos contienen expectativas de la FDA y pensamiento científico actual que no es requerido por la ley. Si pensamos en las regulaciones como lo que la ley exige que hagan los desarrolladores de medicamentos, podemos pensar en los documentos de orientación como un manual de cómo hacerlo en la mayoría de los casos, pero no en todos.
Los documentos contienen expectativas de la FDA y pensamiento científico actual que no es requerido por la ley. Si pensamos en las regulaciones como lo que la ley exige que hagan los desarrolladores de medicamentos, podemos pensar en los documentos de orientación como un manual de cómo hacerlo en la mayoría de los casos, pero no en todos.
Los marcos de orientación son grupos de documentos de orientación relacionados, como los relacionados con la CGT. La creación de nuevas directrices es una parte importante de la misión continua de la FDA de acelerar las innovaciones que hacen que los productos médicos sean más eficaces, más seguros y más asequibles. Los documentos de orientación se redactan y publican de acuerdo con un proceso llamado Buenas prácticas de orientación, que describe cómo el personal de la FDA reunirá los comentarios de expertos y del público en recomendaciones de orientación.
Expectativas crecientes para la CGT en la hemofilia
El primer borrador del documento de orientación de la FDA dedicado al desarrollo temprano de ensayos clínicos de CGT (Guía para la industria: Consideraciones para el diseño de ensayos clínicos de fase temprana de productos de terapia celular y génica) se publicó como borrador en 2013 y se finalizó en 2015. Se discutióA Muchas recomendaciones para la fabricación, pruebas y seguridad y seguimiento del paciente.
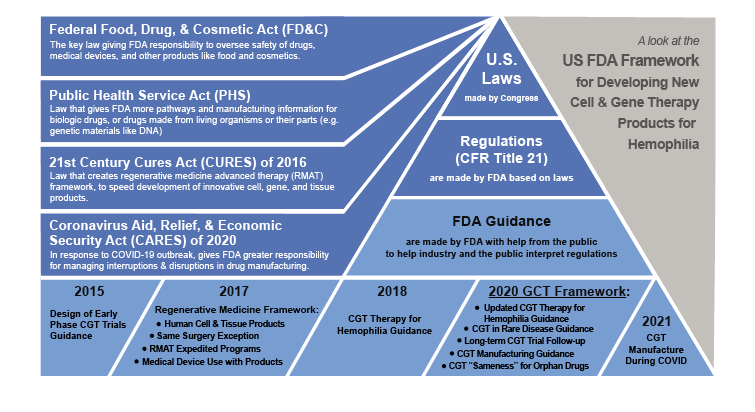
En 2016, el Congreso aprobó la Ley de Curas del Siglo XXI o Ley de Curas, que incluye disposiciones diseñadas para acelerar y agilizar el desarrollo de nuevos medicamentos innovadores. Esta ley se basa en las responsabilidades existentes de la FDA y las terapias de medicina regenerativa establecidas, incluida la terapia génica, la terapia celular, los productos elaborados a partir de tejidos y las combinaciones de estos productos. La FDA publicó una guía en 2017 y la actualizó en 2020 para ayudar a la industria a aplicar estas nuevas regulaciones que en conjunto se conocen como el marco de medicina regenerativa de la FDA. La FDA considera terapias de medicina regenerativa tanto la edición y transferencia de genes humanos como las células modificadas genéticamente que conducen a una producción sostenida de factor en la hemofilia.
La FDA publicó en 2018 una guía para el desarrollo de productos CGT específicos para la hemofilia. Luego, en 2020, se actualizó cuando la FDA lanzó su marco ampliado para terapias celulares y genéticas. Este marco contenía varios documentos de orientación nuevos, incluido el CGT sobre hemofilia. Esto elevó el número total de documentos de orientación en el marco a 27. En 2021, podremos ver cómo la evolución de la terapia celular y genética de la hemofilia se ha visto afectada por el desarrollo de este marco.A
ORIENTACIONES, REGULACIONES Y LEYES QUE CONFORMAN EL MARCO DE LA FDA DE EE. UU. PARA EL DESARROLLO DE NUEVOS PRODUCTOS DE TERAPIA CELULAR Y GÉNICA (CGT) PARA LA HEMOFILIA
Principios de 2020: la FDA lanza un marco de orientación para terapias celulares y génicas
El 28 de enero de 2020, la FDA lanzó su marco de orientación histórico para productos CGT. Las actualizaciones incluyeron mejoras a la guía para la hemofilia y los trastornos raros redactada por primera vez en 2018, incluida la Guía para la industria: Terapia genética humana para la hemofilia y la Guía para la industria: Terapia genética humana para enfermedades raras. La FDA también confirmó el borrador  recomendaciones, Guía para la industria: seguimiento a largo plazo después de la administración de productos de terapia génica humana, para realizar un seguimiento de la seguridad de los pacientes durante cinco a 15 años después de participar en un ensayo clínico CGT. A
recomendaciones, Guía para la industria: seguimiento a largo plazo después de la administración de productos de terapia génica humana, para realizar un seguimiento de la seguridad de los pacientes durante cinco a 15 años después de participar en un ensayo clínico CGT. A
El marco también actualizó la guía de mejores prácticas para que los fabricantes fabriquen y prueben nuevos productos CGT, Guía para la industria: información sobre química, fabricación y control para la terapia génica humana y aplicaciones de nuevos fármacos en investigación. Finalmente, se publicó un nuevo borrador de guía para ayudar a aclarar que los productos que tratan enfermedades raras deben ser únicos (es decir, no “iguales”, como variaciones menores de la misma célula o material genético) para solicitar el estatus de medicamento huérfano, lo que otorga ciertos incentivos como créditos fiscales, exenciones de tarifas de la FDA y derechos para comercializar un nuevo medicamento exclusivamente (o sin competidores que vendan el mismo medicamento) durante siete años después de la aprobación, Guía para la industria: interpretación de la igualdad de los productos de terapia génica según las regulaciones de medicamentos huérfanos. Este marco de orientación aclaró muchos de los problemas que enfrentan los desarrolladores de tratamientos celulares y genéticos para la hemofilia y otros trastornos hemorrágicos poco comunes.
El nuevo panorama para la hemofilia CGT
En 2021, se están llevando a cabo más de una docena de ensayos de transferencia de genes, edición de genes y terapias celulares genéticamente modificadas para la hemofilia, siendo las terapias génicas AAV las más avanzadas, aunque las terapias génicas con lentivirus y las terapias celulares no virales modificadas genéticamente están en curso. El brote de la enfermedad respiratoria COVID-19 causada por el nuevo coronavirus SARS-CoV-2 y la aprobación de la Ley de Ayuda, Alivio y Seguridad Económica por el Coronavirus de 2020 por parte del Congreso han cambiado el enfoque de la FDA. En enero de 2021, la FDA agregó recomendaciones para los fabricantes de CGT a su marco de orientación, Guía para la industria: Consideraciones de fabricación para productos de terapia celular y genética con licencia y en investigación durante la emergencia de salud pública de COVID-19.
CGT para la hemofilia en 2021 y más allá
A pesar de los muchos desafíos que enfrentan los ensayos clínicos en curso en 2020, los tratamientos CGT para la hemofilia están mucho más cerca de llegar a los pacientes. Muchos ensayos clínicos se han beneficiado de una amplia adopción.  de salud digital y telemedicina en 2020, y de la estandarización adicional de las prácticas de fabricación de CGT bajo el marco de orientación actualizado de la FDA.
de salud digital y telemedicina en 2020, y de la estandarización adicional de las prácticas de fabricación de CGT bajo el marco de orientación actualizado de la FDA.
La comunidad CGT ha dado importantes primeros pasos en el camino hacia la armonización o el desarrollo de recomendaciones internacionales aplicables en todo el mundo. Sin embargo, varios ensayos de terapia génica para la hemofilia también han experimentado reveses relacionados con la fabricación o distribución de productos CGT durante el brote de COVID-19 y retrasos en los ensayos clínicos mientras se investiga y comparte con la FDA nueva información científica y de seguridad. Esta comunicación entre los desarrolladores de productos y las agencias reguladoras, como la FDA, es una parte muy importante del desarrollo de tratamientos seguros y eficaces. El brote de COVID-19 también podría aumentar el tiempo que tardan estos tratamientos en estar ampliamente disponibles. Si bien el marco de orientación actual ha ayudado a agilizar el desarrollo de CGT, esperamos que la FDA continúe trabajando estrechamente con investigadores, desarrolladores de productos y grupos de pacientes para seguir evolucionando el marco de orientación de CGT para ayudar a garantizar que la próxima generación de tratamientos para la hemofilia se desarrolle de manera rápida y eficiente. y de forma segura.A


