Si tiene hemofilia A (también llamada hemofilia clásica), le falta o tiene una deficiencia (nivel más bajo) del factor de coagulación VIII (FVIII). Esto significa que su sangre no puede formar un coágulo con éxito. La hemofilia A es hereditaria. Debido a que es una afección ligada al cromosoma X, los hombres se ven más afectados y, por lo tanto, se diagnostican con mayor frecuencia. La hemofilia A afecta a uno de cada 5.000 nacimientos de varones en los EE. UU. Aproximadamente 400 bebés nacen con hemofilia A cada año. Más de 1,1 millones de personas en todo el mundo viven con hemofilia y alrededor de 30 000 viven con ella en los Estados Unidos. Todas las razas y grupos económicos se ven afectados por igual.
La mayoría de las personas con hemofilia A que tienen acceso al tratamiento médico adecuado tienen una expectativa de vida normal y pueden llevar una vida bastante normal.
Una mujer portadora de hemofilia que experimenta síntomas hemorrágicos se denomina portadora sintomática si su nivel es superior a 50% y tiene síntomas hemorrágicos. Las mujeres portadoras que tienen entre 5% y 50% tienen hemofilia leve. La gravedad de la hemofilia A es la misma para las mujeres que para los hombres. Las mujeres con hemofilia y las portadoras sintomáticas tienen los mismos síntomas que cualquier persona con hemofilia. Algunos portadores tienen niveles completamente normales de FVIII y no muestran signos de sangrado.
Si tiene hemofilia A, debe hacerse chequeos regulares con un hematólogo o visitar un Centro de Tratamiento de Hemofilia anualmente. Si usted o alguien de su familia experimenta síntomas de hemofilia A y aún no ha sido diagnosticado, debe comunicarse con su proveedor médico para que lo remita a un hematólogo o HTC para realizar pruebas y diagnóstico.
La hemofilia A suele ser hereditaria y es un rasgo recesivo ligado al cromosoma X, lo que significa que el gen defectuoso se encuentra en el cromosoma X. En casos raros, la hemofilia A puede adquirirse debido a otra situación médica.
Hemofilia A hereditaria
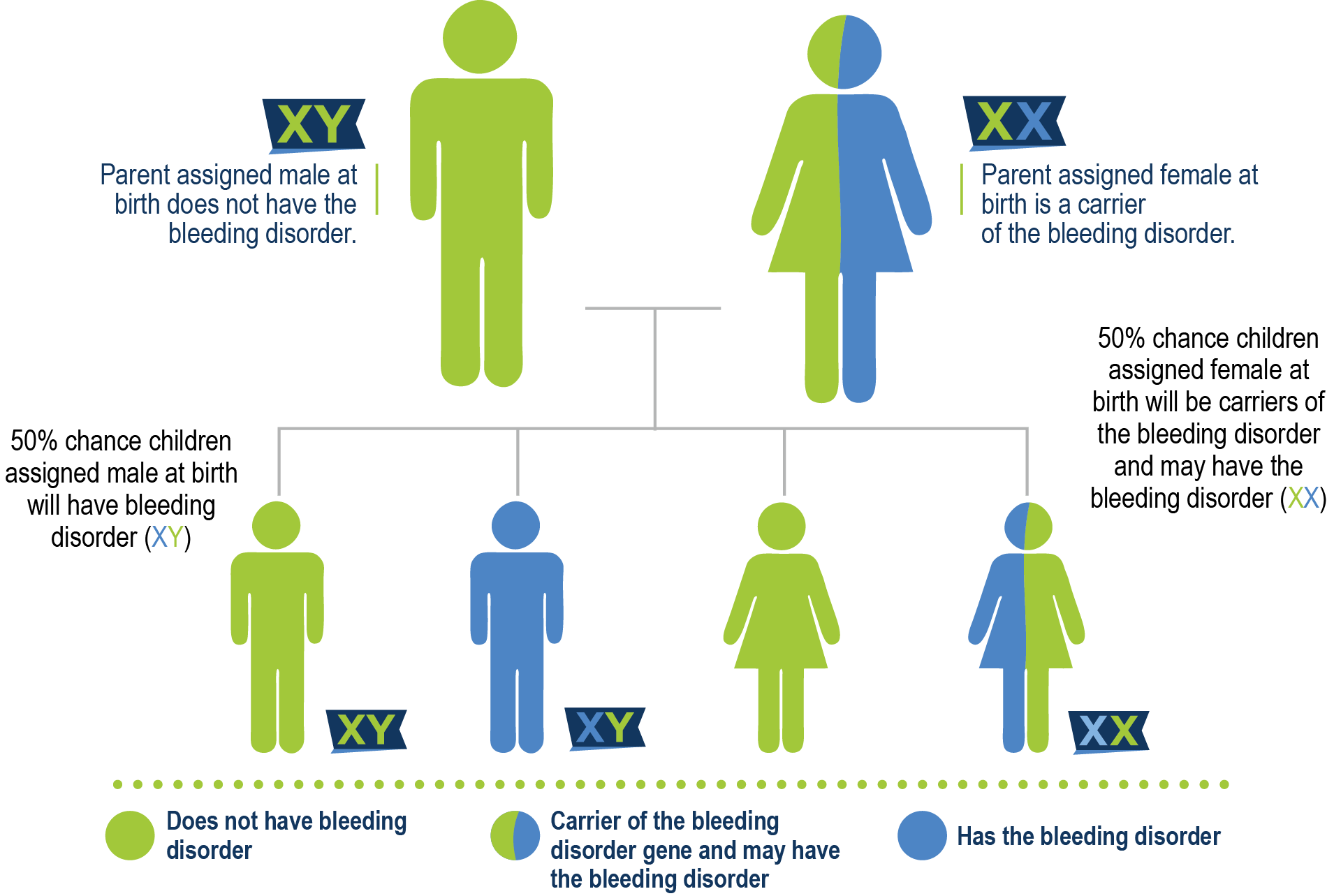
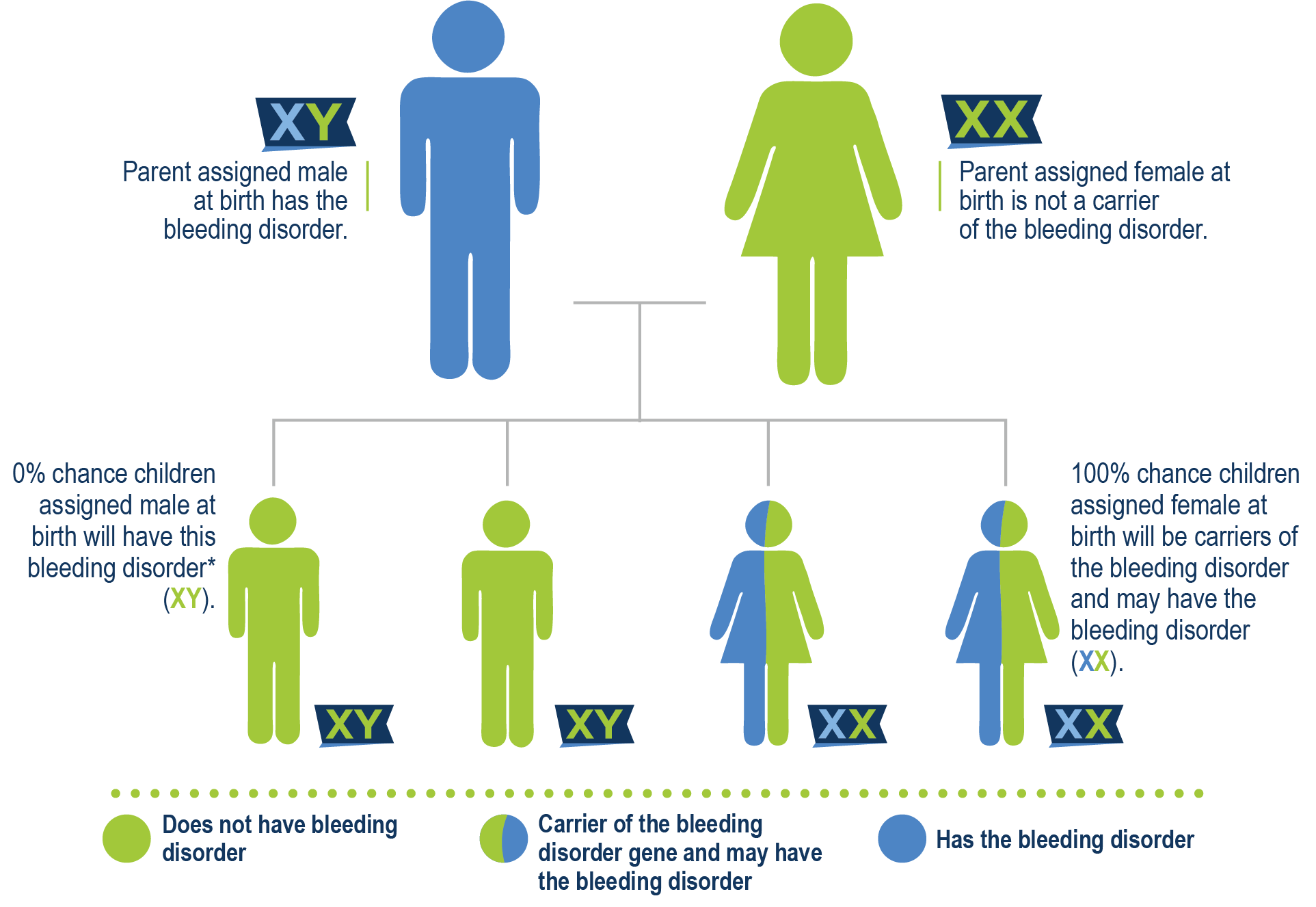
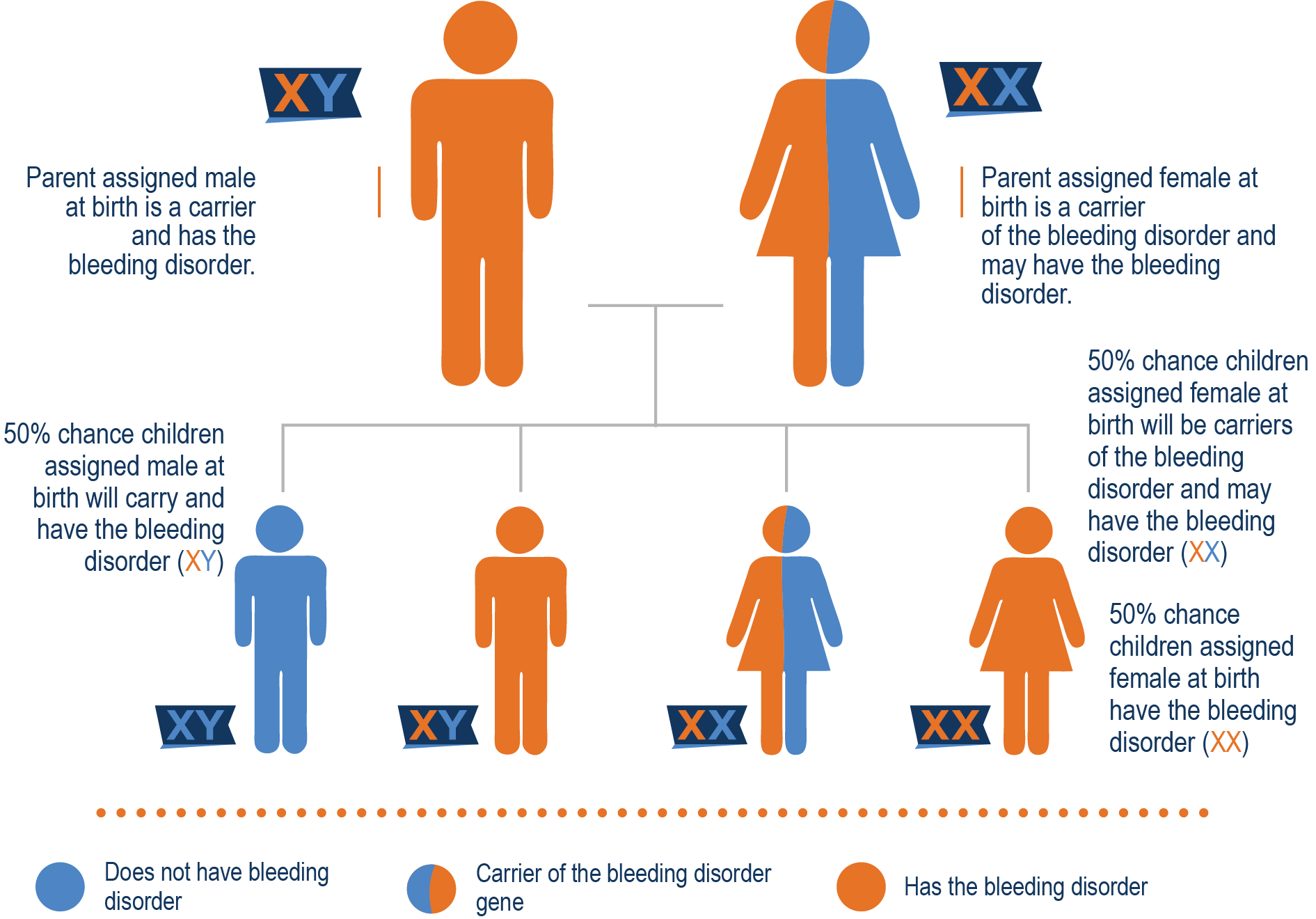
Casi todas las personas que tienen hemofilia A nacen con ella. La hemofilia A no es contagiosa y no se propaga como un virus o una infección. En el 70% de los casos de hemofilia A, hay antecedentes familiares conocidos. El gen que contiene las instrucciones para producir el factor VIII está en el cromosoma X. Cuando ese gen está mutado, puede haber una falta o deficiencia de FVIII. Una madre que porta el gen se denomina portadora y tiene una probabilidad del 50% de tener un hijo con hemofilia y una probabilidad del 50% de tener una hija que también sea portadora. Un padre que tiene hemofilia posee el gen y se lo transmite a su hija porque las hijas reciben dos cromosomas X, uno de su madre y otro de su padre. Por eso, a las hijas de hombres con hemofilia se les llama portadoras obligadas. Dado que los hijos solo reciben un cromosoma Y de su padre, no habrían heredado la hemofilia.
la madre es portadora
Padre tiene hemofilia A
La madre es portadora y el padre tiene hemofilia A
Mutaciones espontáneas
Aproximadamente el 30% de los casos de hemofilia son causados por una mutación espontánea del gen. En algunos casos, la madre no es portadora de hemofilia y el niño está afectado. Este niño puede ser el primero en la familia en tener hemofilia y portar el gen del factor defectuoso. En algunos casos, la mutación genética ocurre en la madre, y ella descubre que es portadora (a través de pruebas de ADN) después de tener un hijo con hemofilia A.
Mujeres Portadoras
Debido a que las mujeres tienen dos copias del cromosoma X, si el gen del factor VIII en un cromosoma no funciona, el gen del otro cromosoma a veces puede hacer el trabajo de producir suficiente factor VIII. Sin embargo, una mujer puede tener una deficiencia genética de FVIII llamada "lionización" o inactivación de X, donde algunas copias del cromosoma X están inactivas. (Esto se llama lionización porque en 1961, una científica llamada Mary Lyon propuso la inactivación aleatoria de un cromosoma X en ratones hembra). Un cromosoma X puede "apagarse" o no expresarse. Si el cromosoma X funcional no se expresa, el gen defectuoso de la hemofilia A toma el control y “ioniza” ese cromosoma X funcional, una mujer portadora puede tener niveles de factor más bajos.
Hemofilia A adquirida
La hemofilia A también puede ser adquirida, causada por una enfermedad, medicación o un trasplante de hígado de alguien que no haya sido diagnosticado con hemofilia. Esto se debe a que el factor de coagulación se produce en el hígado. Adquirir hemofilia A a partir de un trasplante de hígado es extremadamente raro.
Si tiene hemofilia A, es probable que sangre durante más tiempo que alguien sin hemofilia A, y puede sangrar interna o externamente. Las personas con hemofilia A no sangran más que las personas sin hemofilia A; simplemente sangran más tiempo. Los sitios más comunes de sangrado son las articulaciones y los músculos. Los bebés varones sin antecedentes familiares a menudo son diagnosticados cuando son circuncidados. Otros problemas de sangrado generalmente aparecen cuando los bebés comienzan a gatear y caminar. Sin embargo, si tiene hemofilia A leve, es posible que no se le diagnostique hasta mucho más tarde en su vida, posiblemente después de un trauma físico o una cirugía.
Síntomas comunes
- Sangrado en los músculos y las articulaciones, que puede causar dolor e hinchazón.
- hemorragias nasales
- Sangrado prolongado por cortes, trabajos dentales, extracción de muelas o cirugía
- Moretones fáciles (moretones inexplicables)
- Sangre en la orina o las heces
- Hemorragia del tracto gastrointestinal y del tracto urinario
- Sangrado que comienza sin motivo
- Grandes moretones/hematomas
- Menorragia (sangrado menstrual abundante o prolongado: más de cinco días, más de 90 ml de sangrado y/o coágulos más grandes que una uva)
Diagnóstico
Un estudio de coagulación para diagnosticar la hemofilia A podría incluir lo siguiente:
- Tiempo de tromboplastina parcial (PTT)
- Tiempo de protrombina
- Nivel de fibrinógeno
- Recuento de plaquetas
- Actividad del factor VIII sérico
Prueba de portador
Una madre que tiene un hijo con hemofilia y que no tiene antecedentes familiares de la afección puede o no ser portadora de hemofilia, según el lugar donde se produjo la mutación genética. Una madre que tiene un hijo con hemofilia debe considerar hacerse una prueba de ADN para detectar la mutación genética de su hijo para ver si es portadora de hemofilia o si la mutación ocurrió en su hijo. La información genética puede ser importante al planificar embarazos posteriores y puede ser necesario compartirla con otras mujeres de la familia que estén pensando en tener hijos.
Gravedad
La gravedad de la hemofilia A se refiere a la cantidad de factor de coagulación presente en la sangre de una persona. Las personas con hemofilia severa tienden a tener hemorragias más frecuentes, mientras que las personas con hemofilia leve pueden sangrar solo con un trauma importante, un procedimiento dental o una cirugía. Hay excepciones a esto, y no todo el mundo sangra "por los números".
Rangos de nivel de factor
- Niveles de factor normal 50% a 150%
- Hemofilia A leve, 5% a 49%
- Hemofilia A moderada, 1% a 5%
- Hemofilia A severa, menos de 1%
Tratamiento
Los tratamientos para la hemofilia A suelen ser la terapia de reemplazo de factor, que se administra mediante infusión (administrar el medicamento en una vena) o la terapia con anticuerpos biespecíficos, que se administra por vía subcutánea (una inyección justo debajo de la piel).
Para las personas con hemofilia A leve, a veces se puede usar desmopresina (DDAVP) como apoyo a corto plazo. La forma en que funciona es que promueve la liberación de FVIII y factor von Willebrand almacenados de las células endoteliales en el revestimiento de los vasos sanguíneos. No funcionará si tiene hemofilia A moderada o severa porque no puede aumentar algo que no está allí.
Además de los productos anteriores, el sangrado puede tratarse con éxito con ácido aminocaproico o ácido tranexámico. Ambos medicamentos se pueden administrar por vía oral.
Si bien cualquier persona con un trastorno hemorrágico puede desarrollar un inhibidor del factor de coagulación, las personas con hemofilia A desarrollan inhibidores con mayor frecuencia que los demás trastornos hemorrágicos. Los inhibidores son anticuerpos que el sistema inmunológico desarrolla porque percibe el FVIII infundido como una sustancia extraña que necesita ser destruida. Los anticuerpos son proteínas que se comen el FVIII antes de que tenga tiempo de detener el sangrado.
Hasta el 30% de las personas con hemofilia A se ven afectados por inhibidores en algún momento de sus vidas. Los inhibidores generalmente ocurren entre la novena y la quincuagésima infusión de concentrado de factor, pero en casos raros también pueden desarrollarse más tarde en la vida.
Mientras que las personas con hemofilia severa tienen más probabilidades de desarrollar inhibidores, 5% a 8% de las personas con hemofilia A leve o moderada también desarrollan inhibidores. Además del tipo y la gravedad de la hemofilia que tiene una persona, existen otros factores de riesgo:
- Edad/número de exposiciones al factor producto
- Antecedentes familiares de un inhibidor
- Raza/origen étnico (las personas de ascendencia afroamericana e hispana corren un mayor riesgo de desarrollar un inhibidor)
- Mutación genética (hay estudios en curso que indican que el tipo de mutación genética que uno tiene puede indicar un mayor riesgo de desarrollo de inhibidores)
¿Cuáles son los síntomas de un inhibidor?
Los inhibidores destruyen el FVIII infundido, así como el FVIII producido naturalmente por el cuerpo en personas con hemofilia leve y moderada. Si está tratando un episodio de sangrado, siguiendo su protocolo de tratamiento actual, y el sangrado no se detiene, o en realidad puede empeorar, esta es una señal clave de que se puede haber desarrollado un inhibidor. El sangrado más frecuente e inexplicable también puede ser un signo de un inhibidor. Una persona que no tiene un inhibidor sanará de las lesiones o se sentirá mejor después de que se le infunda el FVIII o se complete su plan de tratamiento.
¿Cómo se detectan los inhibidores?
Cualquier persona con hemofilia A debe hacerse la prueba de inhibidores al menos una vez al año. La prueba es una simple extracción de sangre que determina si hay un inhibidor presente. El nivel de inhibidor se denomina título de inhibidor, y eso indica la gravedad del inhibidor. Las dos pruebas utilizadas se denominan ensayo Nijmegen-Bethesda (NBA) y ensayo Bethesda (BA). El título de NBA se mide en unidades Nijmegen-Bethesda (NBU) y el título de BA se mide en unidades Bethesda (BU), pero muchos médicos se refieren a "BU" independientemente de la prueba que se utilice.
¿Todos los inhibidores de la hemofilia A son iguales?
No. Los inhibidores vienen en diferentes grados de severidad. Los inhibidores se clasifican de la siguiente manera:
Título bajo: cuando el nivel de inhibidor en la sangre es inferior a 5 UB. El número, y por lo tanto la fuerza, del inhibidor es bajo. Las personas con inhibidores de título bajo a veces pueden continuar usando productos de FVIII para tratar hemorragias; solo necesitan mucho más. Los inhibidores de títulos bajos a veces pueden resolverse por sí solos.
Título alto: cuando el nivel de inhibidores que se encuentra en la sangre es mayor a 5 UB. El número, y por lo tanto la fuerza del inhibidor, es alto. Las personas con un título alto de inhibidor no se benefician del FVIII, sin importar cuánto se infundan.
Si a usted o a su hijo se les diagnostica un inhibidor, su hematólogo probablemente querrá que no realice la infusión durante 48 horas para verificar su análisis de BU.
Respuesta anamnésica
Una respuesta anemnésica es la producción rápida renovada de un anticuerpo en el segundo (o subsiguiente) encuentro con el mismo antígeno. Una vez que un inhibidor está presente, la fuerza con la que el cuerpo reacciona a una mayor exposición del concentrado de factor, también llamada respuesta inmunitaria o respuesta inmunológica, puede clasificar aún más el tipo de inhibidor.
Respondedor bajo: cuando las personas con inhibidores de respuesta baja reciben FVIII, el título del inhibidor no aumenta. Debido a que el título se mantiene bajo, es posible que puedan controlar el sangrado usando mayores cantidades de concentrados de FVIII.
Alta respuesta: cuando las personas con inhibidores de alta respuesta están expuestas al FVIII, el sistema inmunitario desencadena rápidamente un desarrollo aún mayor de inhibidores. En este caso, se puede usar un agente de derivación para controlar el sangrado.
